PELSA (PEptide-centric Local Stability Assay)是我们最近发表在Nature Methods(https://doi.org/10.1038/s41592-024-02553-7)上的小分子结合蛋白质鉴定方法(图1)。该方法无需对配体进行化学修饰就能实现靶蛋白和结合位点的同时鉴定,灵敏度比同类的LiP-MS技术高一个数量级。PELSA的特点是广谱适用于各种不同结构的配体,而且不依赖于亲和力的大小。PELSA具有自主知识产权,其中中国专利已经获得授权,美国、欧洲等国家和地区的专利也已经申请。该工作从2021年2月5日投到Cell到2024年12月10日发表于Nature Methods历时3年10个月,投稿过程非常艰辛和煎熬。我的学生、也是该工作的第一作者李柯佳, 应计算所贺思敏老师的邀请已经写了一个回顾(附在本文最后),我在这里主要回顾一下当时我为什么觉得应该投Cell,并讨论一下为什么PELSA有比较高的灵敏度以及对于新的分子如何选择合适靶蛋白质鉴定方法。
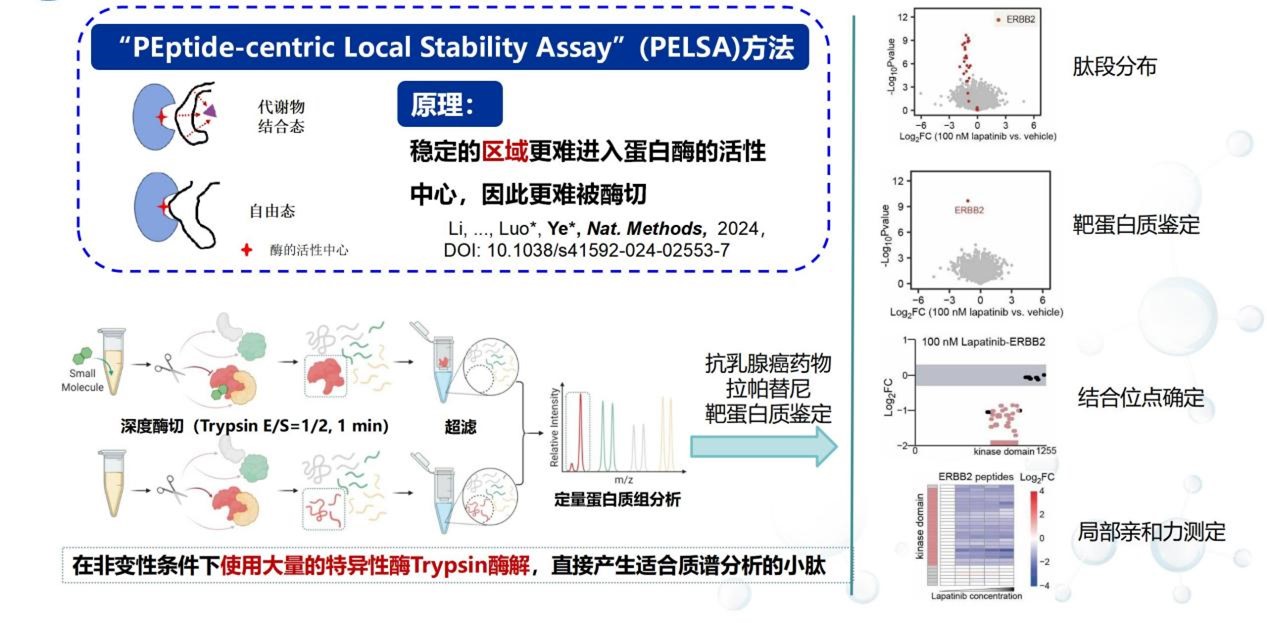
图1 PELSA技术能实现从复杂体系中鉴定配体的靶蛋白质、结合区域以及测定亲和力
在PELSA出现之前,在配体免修饰的靶蛋白质鉴定领域,灵敏度最高、应用最广的是热蛋白质组技术(Thermal Proteome profiling, TPP)。该技术最早由瑞典卡罗林斯卡学院的Par Nordlund教授发展 (Science, 2013, PMID: 23828940)并获得了专利授权。该方法原来的名称是CEllular Thermal Shift Assay (CETSA),主要利用抗体确认活细胞中药物的作用靶蛋白质。后来Mikhail Savitski(现欧洲分子生物学实验室PI)等人将CETSA与质谱技术连用,发展为能在复杂体系中鉴定药物结合蛋白质的靶蛋白质鉴定技术TPP (Science, 2014, PMID: 25278616)。应用于广谱激酶抑制剂星孢菌素靶蛋白质的鉴定时,在要求80%以上为激酶的筛选条件下可以鉴定51个激酶。我们的PELSA方法,开始的时候可以鉴定89个激酶,后来经过优化可以超过100个(见图2),灵敏度与TPP比有明显的优势。此外,PELSA更大的优势在于还能提供结合位点的信息。TPP的优势在于能做活细胞,而PELSA目前主要应用于细胞或组织裂解液。我们PELSA的数据也得到了Nordlund、Savitski两位老师的肯定,并在投稿过程中给了我们很多很好的意见和建议。
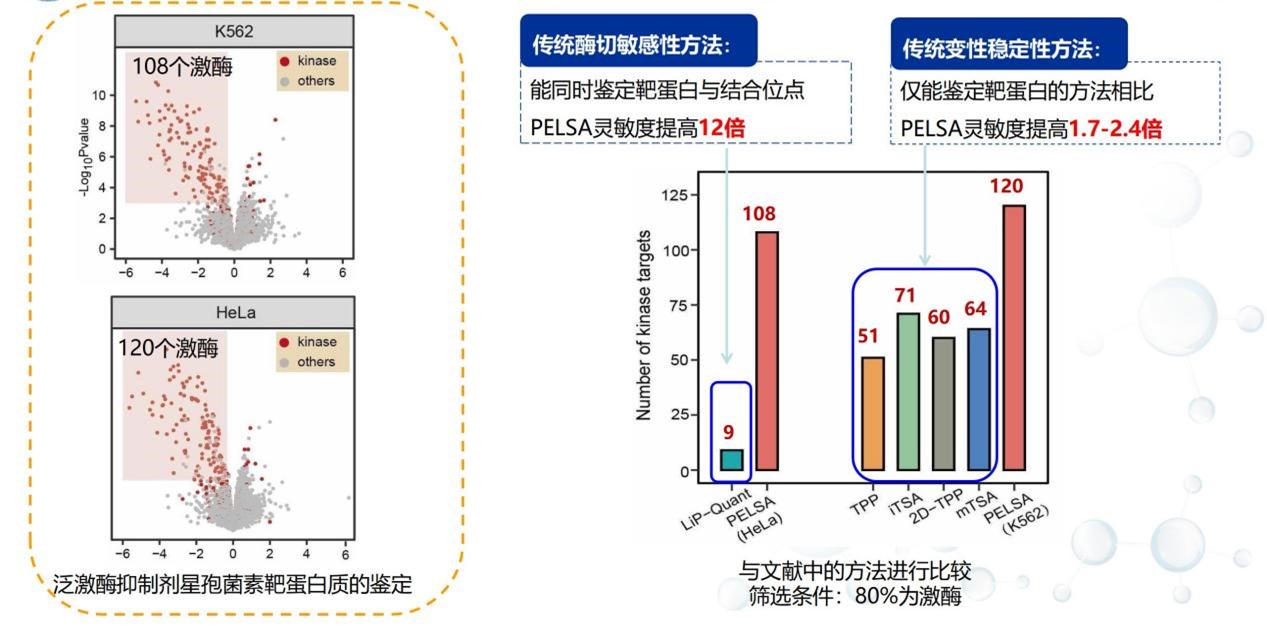
图2 泛激酶抑制剂评估PELSA靶蛋白鉴定的灵敏度
LiP-Quant是LiP-MS技术的改进版 (llaria et al, Nat commun, 2020); TPP指热蛋白质组技术 (Savitski et al, Science, 2014); iTSA, 2D-TPPHE和mTSA 是改进版TPP (Ball et al, Commun Biol, 2020; Zinn et al, J.Proteome Res, 2020; Ruan et al, Anal Chem, 2022)
LiP-MS是由瑞士苏黎世联邦理工学院Paola Picotti教授建立,早期应用于发现蛋白质结构的变化 (Nature Biotechnology, 2014, PMID: 25218519; Science, 2017, PMID: 28232526),后来展示了能在大肠杆菌裂解液中鉴定代谢物结合蛋白质并同时揭示结合位点的能力(Cell, 2018, PMID: 29307493)。我当时对LiP-MS技术很感兴趣,所以也安排学生做了,但是在药物靶蛋白质的鉴定方面效果很差。也有些老师告诉我,他们也做不好LiP-MS。所以当时也有点怀疑LiP-MS在靶蛋白质鉴定方面性能确实比较差。他们Cell的文章也没有与其他方法做系统的比较,性能实际上是很难判断的。LiP-MS此前只做大肠杆菌等简单样品,直到2020年他们终于在真核细胞样品中测试了LiP-MS鉴定药物靶标的性能 (Nature Communications, 2020, PMID: 32826910),结论是:在一个药物剂量下LiP-MS不能区分真的鉴定与假的鉴定(“we realized that this workflow with one drug dose may not discriminate unequivocally true protein interactors and false positive identifications”)。这说明我们原来的预期是对的,LiP-MS在靶蛋白质的鉴定方面性能很差。为了提高药物靶蛋白质的鉴定能力,他们发展了LiP-Quant,主要利用多个药物剂量实验和机器学习来提高性能。在使用7个药物剂量分析星孢菌素靶蛋白质时鉴定了21个激酶,但使用的筛选条件不严格,假阳性很高。如果使用和我们一样严格的筛选标准(80%的蛋白质为激酶),LiP-Quant仅能鉴定9个激酶靶蛋白,而我们的PELSA方法使用一个药物剂量就鉴定到了89个激酶靶蛋白,灵敏度提升了10倍。此后PELSA经过优化后性能得到了进一步提升(图2)。
我们的PELSA技术与LiP-MS技术都是限制性酶切技术,能在鉴定靶蛋白质的同时揭示结合位点,因此属于同类技术。PELSA灵敏度显著高于LiP-MS,即使对比多剂量的LiP-Quant,PELSA也有一个数量级的提升。在与不同类的TPP技术比较,PELSA在鉴定灵敏度方面也有显著提升。因此,我判断PELSA是靶蛋白质鉴定领域的一大突破,应该可以投顶刊,所以我们第一稿投了Cell了,一周后就送审了。在这一稿中我们以药物脱靶鉴定为出口,感谢药物所罗成老师在脱靶蛋白质验证方面的帮助。
这轮审稿被拒后,明显感觉到第1、3个审稿人不太友好,但他们提出的问题都很容易回复。他们让我们做些更具挑战性的应用,所以我们后来重点做代谢物结合蛋白质的鉴定,发现PELSA在这方面优势非常明显。在第二个版本中,以对正常代谢物αKG和肿瘤代谢物R2HG作用蛋白的系统鉴定作为出口。利用PELSA在HeLa细胞裂解液中鉴定到了40个αKG作用蛋白,其中有30个是经过文献验证的已知αKG作用蛋白。2018年Cell文章中LiP-MS的方法在大肠杆菌裂解液中鉴定到了34个αKG作用蛋白,其中仅有2个是已知的αKG作用蛋白。这一实验结果说明PELSA在代谢物靶蛋白鉴定可信度和灵敏度方面,都远超文献报道的LiP-MS方法。我们也发现PELSA在鉴定代谢物等弱相互作用时,灵敏度也比TPP高很多。相关的实验结果可以在我们最终发表的nature methods文章的附件中找到。
那为什么PELSA的配体靶蛋白质鉴定灵敏度能比Lip-MS高一个数量级?实际上两个方法的设计思路是不一样的。LiP-MS鉴定结合位点的设计机理基于靶蛋白质在结合配体后掩盖了一些蛋白质表面的酶切位点,因此使用非特异性酶轻微酶切(图3A)。这种方案需要两步酶切(图3B):第一步使用非特异性的酶在非变性条件下轻微酶切(E/S=1/100),产生比较大的蛋白质片段;为了使该方法与鸟枪法蛋白质组技术兼容,将第一步的酶切产物变性并利用特异性的胰蛋白酶进一步酶切成小的肽段。只有在第一步非变性条件下酶切产生的酶切位点才反映蛋白质的在结合配体后构象的变化,但是含有这些酶切位点的肽段与第二步产生的大量肽段共同存在,使得这些肽段鉴定的效率非常低,因此LiP-MS灵敏度非常低。PELSA方法的设计原理不同(图3A):靶蛋白质在结合配体后,结合区能量降低,稳定性提高,该区域的序列会变的更不容易活动而不容易进蛋白酶的活性口袋。为了使处于低能量区的序列也能酶切,使用高酶浓度(E/S=1:2);为了使产生的肽段更容易被蛋白质组分析,PELSA使用胰蛋白酶(Trypsin)。在高的蛋白酶浓度下,蛋白首先被切为较大的亚基,由于酶切是一个破坏性过程,处于高能量状态或无配体结合的该亚基的结构迅速展开,诱导自身进一步酶切,最终产生拥有两个能反映蛋白区域稳定性酶切位点的,适合自下而上质谱分析的小肽段。而处于低能量状态或有配体结合的该蛋白区域仍然维持稳定结构,较难进入蛋白酶的活性口袋,因此该区域不容易产生小肽段。通过比较此类肽段丰度差异来确定能量状态有差异的蛋白;分析差异肽段在蛋白上的位置,能够确定能量状态有差异的蛋白区域。在该方法中蛋白在非变性的条件下直接产生含有两个能反映蛋白区域稳定性的酶切位点的小肽段,由于小肽段的两个酶切位点距离较近,在能量状态发生变化或有配体结合时,每个位点都将产生差异,两个位点差异的累积使得所属肽段对蛋白能量状态的变化或者配体结合有更大幅度的响应。另外由于该方法对蛋白进行了较为彻底的破坏性酶切,酶切位点不局限于无序区域因此能够产生更多反映蛋白稳定性变化的肽段。再者,该过程仅使用一种蛋白酶,酶切肽段的复杂度大大降低,因此能覆盖到更多的蛋白,以上特点使得该方法具有很高的灵敏度。从上面的讨论可以看出,LiP-MS主要从构象的变化来设计方法,而我们的PELSA主要从局部稳定性这一热力学性质的变化进行设计。PELSA检测的本质是配体结合后局部稳定性的变化。
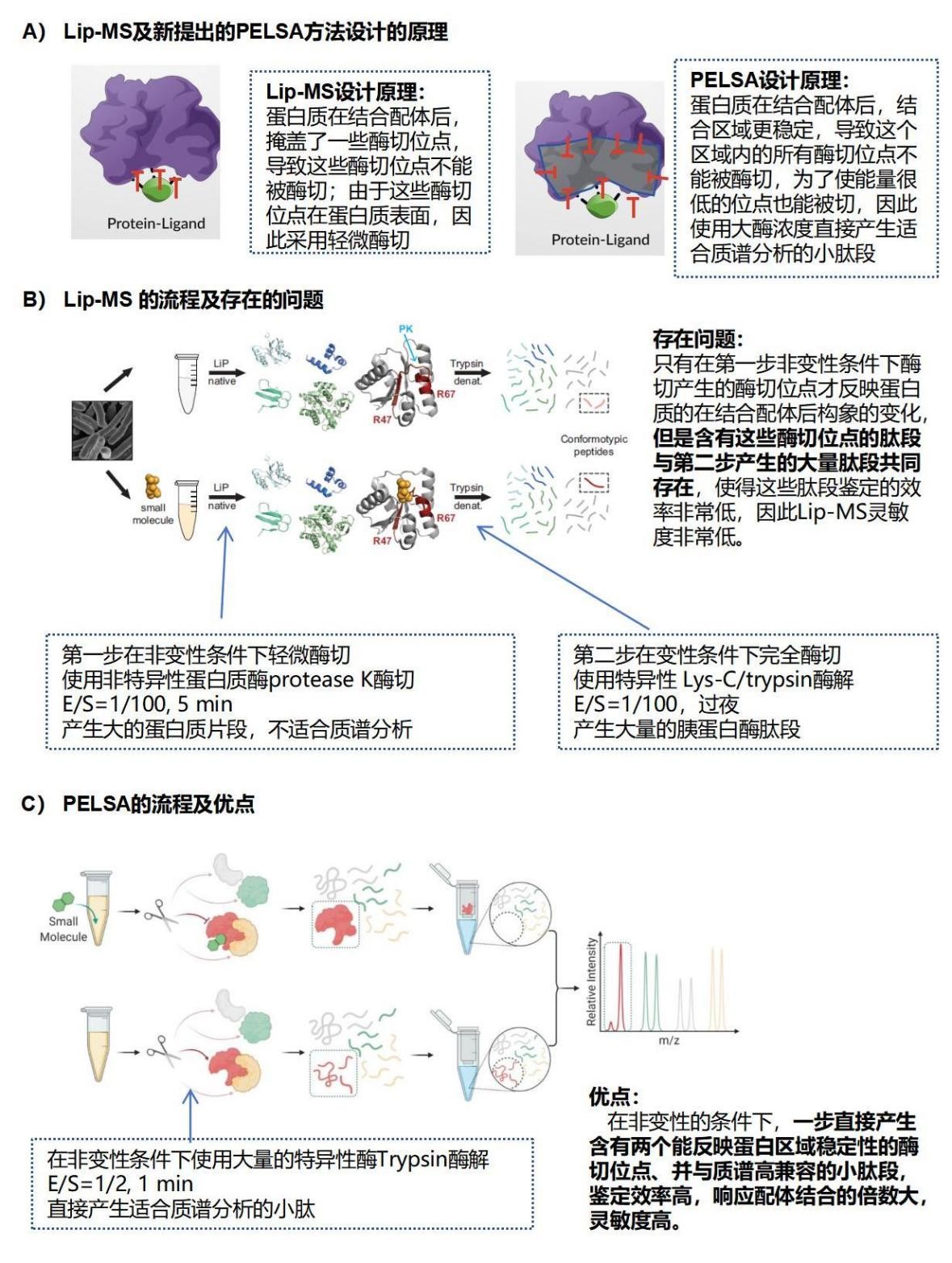
图3 PELSA与Lip-MS的原理及流程对比
这些配体免修饰的方法也存在一些固有的缺点。为了保持蛋白质的天然构象,一般使用不加任何变性剂的缓冲液来提蛋白质,因此提出的膜蛋白质相对比较少,对膜靶蛋白质的鉴定不太友好;这类方法需要直接在细胞裂解液等复杂体系中鉴定靶蛋白质,因此消耗的质谱时间比较长,也不容易鉴定低丰度蛋白质。经典的亲和富集方法,通过改造配体合成亲和探针,利用探针将结合蛋白质从复杂的蛋白质样品中富集出来,然后进行质谱分析。这种方法的优点是得到的样品很简单,消耗的质谱时间比较短;而且由于没有高丰度蛋白质的干扰,更容易鉴定低丰度蛋白质。基于探针方法的一个主要缺点是,在共价改性配体时容易导致配体的结合性质改变。因此,为了获得对配体结合影响比较小的探针,需要用户有很强的有机化学的知识并做大量的活性测试,因此很费时费力。对有些配体甚至找不到合适的方法合成亲和探针。另外,这类方法不适用于弱相互作用靶蛋白质的鉴定。从上面的分析可以看出,如果潜在的靶蛋白质丰度很低或者是膜蛋白质,利用PELSA、TPP等免修饰的方法进行靶蛋白质的鉴定成功的机率相对较小。如果对于一个完全未知的化合物,建议先使用PELSA方法进行尝试,不成功再使用其它方法。如果是弱相互作用,如代谢物结合蛋白质的鉴定,PELSA应该是目前最好的选项了。
我们的PELSA文章没有深入的功能研究,但是利用各种各样的配体,包括药物、金属离子、代谢物、抗体和翻译后修饰肽段等,充分证明了方法的优越性和广谱适用性。有些高档次文章则胜在故事比较精彩或是它的功能比较新,但是方法的性能不一定很好。我们实验室在药靶鉴定领域积累了一些经验。如果让我们来评价靶蛋白质鉴定方法的性能,我们会在相同的条件下用使用相同的多靶点药物进行系统评价。强相互作用可以选广谱激酶抑制剂星孢菌素,弱相互作用可以选αKG 。一个新手来尝试PELSA实验,也建议先鉴定星孢菌素的靶蛋白质。在跟我们相似的条件下,能鉴定到90个以上的激酶,就基本说明实验步骤没有大问题。
我们PELSA方法,是以肽段为中心的局部稳定性的分析方法,涉及大量肽段定量数据的处理和展示,数据处理相对比较复杂。我们近期将会发布一个专门用于PELSA数据处理的软件PELSA-Decipher,将能非常方便的进行数据的统计、结合区域的展示以及亲和力的求算。相关信息将会发布在我们的微信公众号上,请大家关注(在微信公众号中搜索PELSA就能找到我们的公众号:PELSA靶标分析)。
叶明亮
2025年2月9日于大连
