主要从事分析化学新方法及其在生命健康领域的应用研究。课题组以色谱分离与质谱分析为立足点,针对蛋白质组等复杂生物样品的高效分离与高灵敏检测问题,开展色谱、质谱分析新技术和新方法及其在生物医学中的应用的研究,注重解决疾病蛋白质组学研究所面临的技术瓶颈问题,让精准医学更精准。课题组在蛋白质组学分析,尤其是翻译后修饰蛋白质组学分析、药物靶标蛋白质鉴定等方面建立了多种独创的分析方法。
主要研究方向:
1)蛋白质的质谱分析新技术:发展基于液相色谱串联质谱(LC-MS/MS)的蛋白质分析、鉴定新方法;发展高精度质谱确定多肽序列的De Novo技术;发展蛋白质质谱分析的样品预处理技术。
2)高效分离柱技术与多维分离分析系统:发展蛋白质与多肽的高效分离方法、新型多维分离模式。
3)蛋白质组修饰谱的分析新方法:发展蛋白质磷酸化、甲基化、糖基化等蛋白质翻译后修饰的分析新方法,包括修饰蛋白和多肽的富集技术、修饰肽段的通量化分离和鉴定技术等;发展修饰蛋白质的谱图解析方法和质谱数据处理方法。
4)药物靶标蛋白质的鉴定技术:发展基于能量状态差异的药物靶标蛋白质的鉴定方法,包括基于蛋白质变性稳定性和酶切稳定性的方法。
5)生物医学应用研究:药物靶蛋白筛选、耐药机制、新型肿瘤生物标志物鉴定等方面的研究。
主要代表性工作介绍:
1)蛋白质翻译后修饰的富集材料与方法
翻译后修饰是蛋白质功能的调控者。课题组在蛋白质磷酸化、糖基化、甲基化等方面发展了一系列新技术、新方法。在蛋白质磷酸化方面,发展了原创的磷酸肽富集新材料(固定钛离子亲和色谱,Ti4+-IMAC)(J. Proteome Res., 2008, 7(9), 3957,Nat. Protoc., 2013, 8(3), 461),由于具有很高的富集特异性,在国内外获得了广泛的应用。为了改善传统的利用Ti4+-IMAC微球进行磷酸肽富集时操作繁琐、不易于实现样品批量化处理的问题,近期发展了一种大粒径的Ti4+-IMAC材料(J. Chromat. A., 2017, 1498, 22-8),将其填充于离心小柱中实现了固相萃取模式下磷酸肽的富集,操作简便,省时省力。将其应用于HeLa细胞的磷酸化蛋白质组分析,富集特异性高达99%。2018年该富集材料在百灵威科技有限公司销售(产品编号: 2749380,2749381),进一步促进了该技术在国内外的应用。将Ti4+-IMAC与SH2超亲体连用,建立了酪氨酸磷酸化肽段的高特异性富集方法,从9个细胞系中高可信地鉴定到了10,030个酪氨酸磷酸化位点,构建了迄今为止最大的酪氨酸磷酸化蛋白质组数据集,体现了酪氨酸磷酸化分析的最高水平(Nat. Chem. Biol., 2016, 12(11), 959 ; Anal. Chem., 2017, 89(17), 9307; J. Proteome Res., 2018, 17(1), 243 ;Cancer Let., 2020, 475(10), 53)。
在蛋白质糖基化方面,建立了比较完善的糖基化分析新方法。在O-GlcNAc糖基化方面,提出并构建了糖肽的可逆酶促化学标记策略,并与亲水作用色谱富集方法相结合,实现了O-GlcNAc糖基化的高灵敏度检测(Angew. Chem., 2022,134, e2021178)。在O-GalNAc糖基化方面,发展了完整O糖肽的检索鉴定新方法(Anal. Chem. 2019, 91, 3852; Bioinformatics, 2022, 38, 7, 1911),通过将糖组成可变修饰设置在肽段层次,有效降低了检索空间,被认为是“to combat this search time issue”的好方法(Nature Methods, 2020, 17, 1133)。在N糖基化方面,开发了一款具有高灵敏度的N-糖肽质谱谱图解析新软件——Glyco-Decipher,可实现在解析谱图的过程中不依赖糖库,利用不同糖肽的同一肽段骨架具有相似碎裂规律的特点,发展出基于“模式识别”的肽段序列鉴定新方法,实现谱图拓展,从而提高了完整糖肽的鉴定灵敏度,并且可发现未知的糖链及糖链修饰 (Nat. Commun., 2022, 13, 1900)。
在蛋白质甲基化方面,发展了首个精氨酸甲基化的化学富集新方法。蛋白质甲基化与表观遗传、肿瘤的发生发展酶切相关,由于甲基化对肽段物理化学性质的影响很小,传统的免疫亲和富集具有特异性低、成本高等缺点。精氨酸二甲基化会影响一些神经退行性疾病相关蛋白的液—液相分离,以及相分离所驱动的无膜细胞器的产生。然而,受限于目前精氨酸二甲基化蛋白质组分析技术覆盖率不足,这类研究只聚焦于少数几个蛋白,尚未有系统性研究过精氨酸甲基化对蛋白质相分离的影响。将硼酸化学引入到甲基化蛋白质组分析方法中,并巧妙利用了精氨酸残基上不同修饰基团的位阻差异,建立了首个精氨酸二甲基化肽段的化学富集方法,显著提高了蛋白质甲基化的分析能力;利用此新方法,系统分析了蛋白质分相过程中精氨酸二甲基化的变化,揭示了精氨酸二甲基化对蛋白质液—液相分离具有重要的调控作用(PNAS, 2022, 119, 43, e2205255119)。
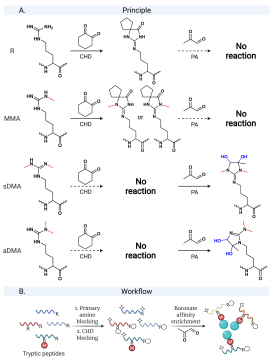
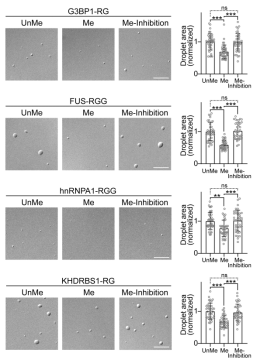
图1 建立了精氨酸二甲基化肽段的首个化学富集方法并应用于蛋白质相分离的研究
2)高稳健位点特异性糖型分析系统的构建及其用于肿瘤标志物的筛选
蛋白质糖基化在蛋白质稳定性、细胞内和细胞间信号转导、激素活化或失活、免疫调节等方面起着重要作用,而蛋白质糖基化的异常表达也通常和遗传病、炎症和癌症等多种疾病的发生发展密切相关。目前,几乎所有用于临床诊断的疾病生物标志物,如甲胎蛋白(AFP)、癌抗原125(CA125)、癌胚抗原(CEA)和前列腺特异性抗原(PSA)等,都是糖基化蛋白质。这些生物标志物的存在说明肿瘤中蛋白质糖基化表达的紊乱,但是它们的特异性和敏感性都还比较低。
蛋白质糖基化是一种非常复杂的蛋白质翻译后修饰,其特点是高度异质性,即一个蛋白质有多达几十个糖基化位点,一个位点能修饰多达百余种糖型。现有在临床上使用的生物标志物要么只检测蛋白质的浓度,要么只检测某类糖型的浓度(如CA19-9就是一类糖抗原),还没有出现基于特定位点糖型的生物标志物。鉴于蛋白质的糖基化修饰与肿瘤的发生密切相关,在组学层次进行位点特异性糖型的分析有望发现新型疾病标志物,提高基于蛋白质糖基化的精准医学研究水平,但其分析仍然面临严峻挑战。
传统的N-糖基化蛋白质组技术要求将糖肽上连接的N-糖链去除之后再进行质谱检测,因此该方法只能鉴定糖基化蛋白质及其位点,而损失了糖链甚至特定位点糖链的信息。针对传统方法的这一缺点,为了实现蛋白质、糖基化位点和特点位点糖链的同时分析,我们建立了一种高稳健的糖基化蛋白质组分析系统。该系统由高重现的自动化完整糖肽富集方法、高稳健的微升流速液相色谱串联二级质谱检测技术、高灵敏度的谱图解析软件等环节组成,能够应用于规模化临床样本的糖基化蛋白质组分析,并筛选新型的位点特异性糖型生物标志物。
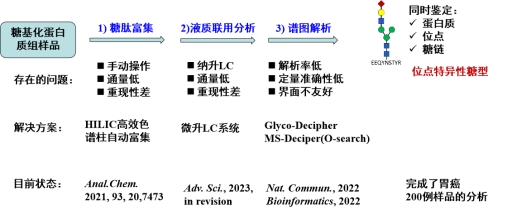
图2 构建了适用于大样本分析的高稳健位点特异性糖型分析系统
在自下而上的糖基化蛋白质组学研究中,复杂生物体系糖基化肽段的丰度远低于其他多肽。因此,糖基化肽段的富集是提高检测灵敏度和可信度必不可少的实验流程。针对传统糖基化蛋白质组学中富集完整糖肽的实验步骤非常繁琐而难以应用于分析大规模临床样品的问题,我们发展了一种自动化完整糖肽富集方法(Anal. Chem. 2021, 93, 7473)。该方法一方面利用亲水作用色谱能够无偏见的保留几乎所有完整糖肽的特点,实现了对生物样品中完整糖肽的富集;另一方面利用超高效液相色谱仪连续自动进样和梯度洗脱的特点,实现了富集流程的自动化;实现了对血清位点特异性N-糖基化蛋白质组学的高富集特异性和高糖肽定量重现性分析,并应用于探究胰腺癌患者血清中蛋白质N-糖基化的变化。该方法是一个能够用于微量复杂生物样品糖基化蛋白质组学分析的强大工具,并且在新型疾病标志物的筛选方面有巨大的潜力。
在蛋白质组学的LC-MS/MS分析中,纳升流速液相色谱通常被用于分离肽段样品,但该技术在规模化分析中存在超压和喷雾不稳定等缺点,因而难以应用于分析大队列样本的糖基化蛋白质组。针对以上问题,我们建立了一种高稳健的微升流速液相色谱串联二级质谱分析方法,应用于分析临床样本的完整糖肽,提高了规模化的糖基化蛋白质组中数据采集的稳健性。通过连续10天和超过200个临床血清完整糖肽样本的数据采集,该方法表现了较强的保留时间稳定性、喷雾稳定性和抗污染能力等特点,并且有高稳定性的完整糖肽鉴定数目(RSD < 5%)以及高重现性的完整糖肽定量结果(Pearson correlation > 0.9)。
由于完整糖肽的质谱谱图高度复杂,目前各糖肽解析软件的谱图解析率低。针对常规糖肽解析策略依赖糖库而无法实现未知糖链及修饰糖的鉴定的问题,我们开发了非糖库依赖的肽段序列鉴定方法,实现了未知糖链肽段及其上可能带有的修饰基团的鉴定。为解决N-糖肽质谱谱图解析率低的问题,我们通过系统研究糖肽的碎裂规律,发现糖链的种类、组成、母离子价态等对肽段骨架的碎裂模式没有显著的影响,并建立了肽段序列相同的完整糖肽谱图之间的联系,发展了基于“模式识别”的肽段序列鉴定策略,实现了完整糖肽的谱图拓展,在原有基础上将完整糖肽的解析率提升了31%(Nat. Commun., 2022, 13, 1900)。通过对同一个N-糖肽质谱数据进行对比分析,发现Glyco-Decipher的谱图解析效率比其它软件提升了34-179%。该软件具有友好的用户界面和较好的定量比较功能,学术界可以免费使用(软件可从GitHub 下载)。
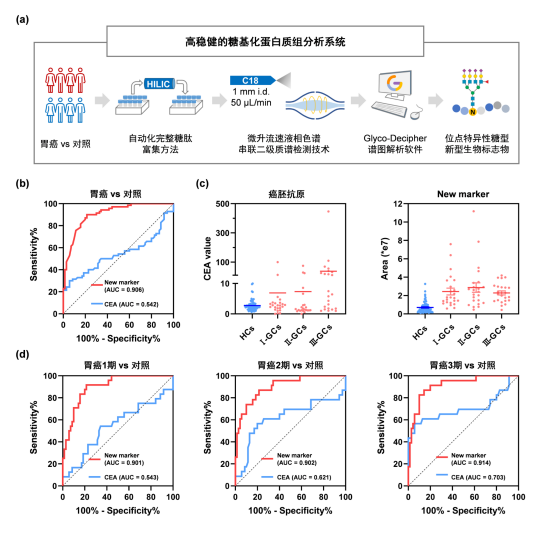
图3 发现了对胃癌具有早诊作用的位点特异性糖型标志物
胃癌是全球第五常见诊断和死亡率第三的癌症,主要原因是早期胃癌无明显症状并且存在较长的潜伏期导致的诊断迟、转移快和治疗效率低。在中国,超过80%的胃癌患者被诊断时已为胃癌晚期,导五年生存率只有35.1%。目前,胃癌诊断主要依赖于内窥镜和活检,这种侵入式的诊疗方式给患者带来较多的疼痛,同时也会因为早期胃癌临床症状不明显导致漏诊,不易于胃癌的快速和高效诊断。而血液、尿液和胃液等体液中存在癌症相关的基因、蛋白质和代谢物等,检测这类基于体液的标志物属于非入侵式的诊断方式,有较好的应用前景。因此,我们通过自动化完整糖肽富集方法、微升流速液相色谱串联二级质谱分析技术以及N-完整糖肽谱图解析软件,构建了一种高稳健的糖基化蛋白质组分析系统,并将该系统应用于研究大队列胃癌患者血清中的N-糖蛋白质组。通过定量糖基化蛋白质的差异分析和构建机器学习随机森林模型,我们发现了一组新型位点特异性糖型生物标志物,对胃癌患者以及早期胃癌患者具有良好的诊断效果。其中,最具代表性的新型生物标志物为四天线末端有唾液酸路易斯抗原的位点特异性糖型,它诊断早期胃癌的受试者工作特征(ROC)曲线的曲线下面积(AUC)约为0.9(特异性,90%;灵敏度,78%),而目前临床使用的经典的胃癌标志物血清CEA诊断早期胃癌的AUC值约为0.6(特异性,60%;灵敏度,80%),并且在相同特异性(80%)条件下,该位点特异性糖型的灵敏度(> 80%)明显优于血清CEA(< 40%)。
3)药物靶标蛋白质的鉴定及其用于药物作用机制的揭示
药物靶标蛋白的系统鉴定有助于降低新药研发成本并揭示药物作用机制及可能的毒副作用。近年来,课题组在基于免修饰药物靶标蛋白质组学鉴定方面发展了一系列方法(Chem. Sci., 2022, 13, 12403; Anal. Chem., 2022, 94, 3352; Anal. Chem., 2022, 94, 6482; ACS Chem. Bio., 2022, 17, 252; Anal. Chem., 2020, 92, 1363; Proteomics, 2020, 20, 1900372; Anal. Chem., 2020, 92, 13912)。建立的基于溶剂沉淀蛋白药物靶标筛选的SIP新方法(Anal. Chem., 2020, 92, 1363)特异性高,操作简单,克服了传统基于药物修饰的化学蛋白质组学方法需要对配体进行修饰的难题,以及热变性方法难以实现部分靶蛋白质对热不敏感的问题,是目前唯一能与热变性方法媲美的方法。该方法具有自主知识产权,中国专利已经授权(授权公告号:CN 112824905 B)并申请了国际PCT专利(专利号:PCT/CN2020/130079)。受邀为 Springer Nature 的《分子生物学方法》书籍撰写SIP方法的具体流程。哈佛医学院Gygi团队认为我们的SIP方法“provided the basic framework for a robust target engagement approach”,并将SIP与TMT标记试剂结合后发展了SPP方法(eLife, 2021, 10, 70784)。我们应用SIP方法成功筛选了紫草素的结合蛋白NEMO/IKKb,紫草素通过破坏这两个蛋白的相互作用从而抑制癌细胞的增殖(图4)。相关工作发表在肿瘤学靶向治疗领域值权威杂志(STTT,2022, 7, 71; IF=38.104)。另外,团队还发展了pH 依赖蛋白沉淀法(pHDPP)(Chem. Sci., 2022, 13, 12403),该方法特异性高、通量高、成本低、操作简单等优势,在靶标鉴定方面与热变性具有互补性。应用pHDPP方法成功筛选到了双青蒿素的候选靶标ALDH7A1和HMGB1(图4)。
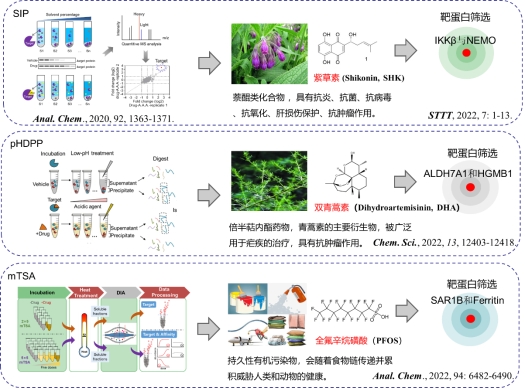
图4 建立了多种免修饰的药物靶蛋白质鉴定新方法并应用于药物靶标的鉴定
为了提高了特异性和灵敏度,克服了TPP方法中仅分析可溶蛋白的局限性,团队发展了基于沉淀辅助的PSTPP方法(ACS Chem. Bio., 2022, 17, 252),该方法同时分析可溶和沉淀部分的蛋白获得丰富的靶标筛选信息。另外,发展的基于深度学习的新型图像识别算法取代了TPP方法中曲线拟合策略,避免了对具有多个转换的部分蛋白质组的歧视。为了能够在蛋白质组水平识别药物结合靶标和结合位点,发展了一种赖氨酸反应性分析方法(Anal. Chem., 2022, 94, 7, 3352)。
针对目前已有的二维蛋白质热迁移分析方法的实验流程复杂、分析通量较低的缺点,结合数据非依赖采集(DIA)模式,开发了一种矩阵热迁移分析方法(mTSA)用于快速地解析药物分子的靶蛋白和结合亲和力(图4)。利用该方法对全氟辛烷磺酸(PFOS)的靶蛋白空间进行解析(Anal. Chem., 2022, 94, 6482)。PFOS是一种持久性有机污染物,会随着食物链传递并进行累计,最终威胁人类和动物的健康,包括影响脂肪酸代谢等。由于PFOS的特殊结构,不合适使用基于化学衍生的化学蛋白质组学方法对其靶蛋白进行解析。在最终的mTSA筛选结果中,在5000个背景蛋白中鉴定到了39个潜在的PFOS靶蛋白,这些靶标蛋白富集于GTP结合蛋白(SAR1A和SAR1B)以及铁离子蛋白(FTH1和FTL)。鉴于得出的PFOS与铁离子蛋白之间的亲和力在0.3μM,远远高于其类似物脂肪酸与铁离子蛋白之间的结合亲和力。因此,推测出了一种PFOS影响脂肪酸代谢的机理,即PFOS可以竞争性地结合在铁离子蛋白上的脂肪酸结合口袋内,将脂肪酸排出,从而影响脂肪酸的代谢过程。
