PELSA is a patented technology we developed to identify ligand target proteins and binding sites at proteomics scale (Li et al., Nat. Methods, 2025; Patent in China is granted, Patents in USA, and other territories are pending; Please contact Prof. Ye for commercial use).
The biochemical functions of proteins invariably involve interactions with ligands of some type, which act as enzyme substrates or inhibitors, signaling molecules, allosteric modulators, structural anchors, etc. Monitoring protein-ligand interactions is thus essential for comprehending various aspects of life science, including drug mechanisms of action, regulatory processes in cellular metabolism and signaling, and the functions of uncharacterized proteins. Additionally, knowledge of the ligand-binding regions holds immense value for structure-based drug design and biological hypothesis generation.
Modification-based methods which rely on chemical modifications of the ligands to capture ligand-binding proteins and binding regions require extensive chemical synthesis and may be not applicable for ligands that lack suitable sites for chemical modification. Previously reported modification-free methods, including CEllular Thermal Shift Assay (CETSA) and Thermal Proteome Profiling (TPP) bypass the need of ligand modification but do not support the identification of specific ligand-binding regions in target proteins. Paola et al. developed LiP-MS (limited proteolysis coupled with mass spectrometry), which can identify ligand-binding proteins and binding regions in the cell lysates of microbial organisms. Despite the advancements brought by LiP-MS and the subsequently developed LiP-Quant (a dose-response version of LiP-MS tailored for complex human cell lysates), their capacity for target identification remains limited. Therefore, an efficient modification-free approach to systematically determine ligand-binding proteins and binding regions from complex proteome is still lacking.
Recently, we proposed extensive trypsinization to directly generate MS-detectable peptides from native proteins to represent protein local stability. This digestion scheme in couple with a simple separation procedure largely reduces the complexity of peptide samples and, crucially, amplifies the readout of ligand-induced protein local stability shifts. Based on this observation, we established a method we term PEptide-centric Local Stability Assay (PELSA) that enables sensitive identification of target proteins while also preserving extensive binding-region information. We demonstrated that PELSA achieves unprecedented sensitivity in revealing ligand-binding proteins through extensive comparisons against alternative methods. Compared against existing modification-free methods that enable binding region determination (LiP-MS methods), PELSA identified 6-fold more FKBP family target proteins (6 versus 1) for rapamycin and 12-fold more kinase targets (108 versus 9) for a pan-kinase inhibitor (staurosporine) than LiP-MS and LiP-Quant, respectively. Compared with prevalent modification-free methods that do not yield binding region information (TPP methods), PELSA identified 1.7-2.4 times more kinase targets for staurosporine than TPP and recently revised TPP methods (iTSA, 2D-TPP, and mTSA).
We also showcased the capacity of PELSA for sensitively and informatively probing weak interactions by identifying the binding proteins of leucine, folate, αKG, and R2HG. While previous studies have employed modification-based or modification-free methods to investigate metabolite-binding proteins, these approaches often generate a large number of candidate targets but with a limited number of known metabolite-binding proteins. In contrast, PELSA results consistently exhibit a significantly higher percentage of known-binding events. For instance, in a prior LiP-MS study of αKG-treated E.coli lysates, 34 candidate targets were identified with 2 known αKG binding proteins. In comparison, PELSA identified 40 candidate targets, and notably, 30 of these were known αKG binding proteins, despite using a more complex lysate sample (human HeLa cell lysate). We envision that the high confident hits obtained by PELSA’s elevated hit rate could significantly facilitate the validation process in hypothesis-generation studies.
In summary, we demonstrated PELSA is a highly sensitive and generic method to reveal binding regions on proteins of very diverse ligand types (including drugs, antibodies, phosphorylated peptides, metal ions, and metabolites) on a proteomics scale, without the need for chemical modification of the analyte ligand. Beyond ligand binding, the transition of a protein between different proteoforms (e.g, the presence or absence of post-translational modification) may also induce protein stability shifts, and thus could also be investigated by PELSA. We envision that PELSA will find wide utilization throughout life science research.
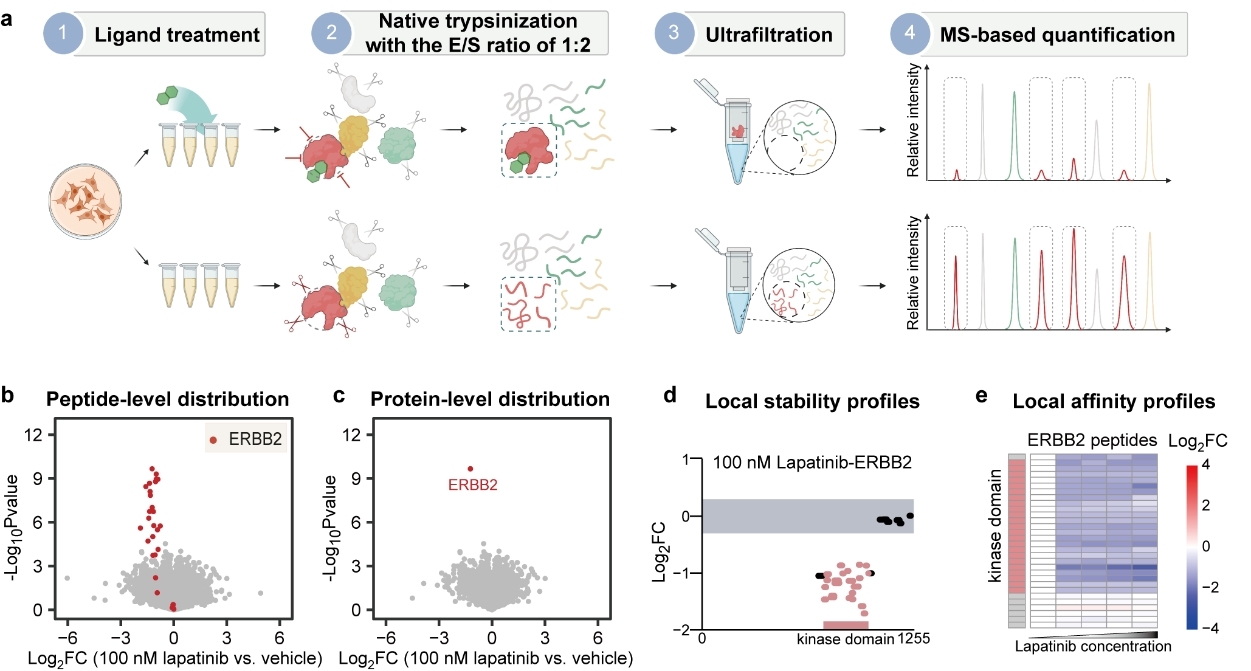
PELSA enables proteome-wide determination of ligand target proteins and binding sites.
a, Workflow of PELSA. E/S ratio refers to enzyme /substrate ratio (wt/wt). b, Volcano plot visualization of all peptides from a PELSA analysis of BT474 lysates exposed to 100 nM lapatinib. c, Volcano plot as in (b) but on the protein level. d, Local stability profiles to reveal ligand-binding regions. The upper and lower boundaries of the grey-shaded area represent log2FCs of 0.3 and -0.3, respectively. e, Local affinity profiles to reveal the local binding affinity of a ligand. Heat map representation of log2 peptide fold changes of ERBB2 with increasing lapatinib concentrations (0 nM, 100 nM, 1 μM, 10 μM, and 100 μM).